动脉粥样硬化是多种心脑血管疾病的病理基础。通过探讨动脉粥样硬化的病因及发病机制发现有效的防治措施,从而延迟甚至逆转动脉粥样硬化病变的进展,降低心脑血管疾病的发病率和病死率,仍是心血管疾病研究领域最重要的课题之一。
早在约100年前就有学者提出感染可能是为动脉粥样硬化的危险因素,但直到1970年才有文献报道支持病毒为动脉粥样硬化病源的学说。
有关动脉粥样硬化的炎症学说认为:慢性炎症在动脉粥样硬化的发生发展中起重要作用。该学说认为动脉粥样硬化是一种慢性炎症性血管疾病,免疫反应造成的血管损伤是动脉粥样硬化发生的中心环节,高脂血症只是动脉粥样硬化发生的始动因素之一。免疫炎症损伤引发动脉粥样硬化的学说日益受到重视。有学者甚至提出动脉粥样硬化是免疫性疾病,而引起免疫反应的抗原可能为自身抗原(如ox-LDL)和病原体(如肺炎衣原体),其中病原微生物感染可引起炎症级联反应,造成免疫损伤,病原微生物在动脉粥样硬化发生和发展中的作用和机制日益受到关注。
有人提出许多病原体感染与动脉粥样硬化有关,如巨细胞病毒(Cytomegaoviyns, CMV)、1型和2型单纯疱疹病毒(Herpes simplex virus,HSV)、肺炎衣原体(Chlamydia pneumoniae, CP)、幽门螺杆菌(Helicobacter pylori, HP)、甲型肝炎病毒以及牙周病相关微生物等。但是,最近几项大规模流行病学研究的阴性结果似乎并不支持感染与动脉粥样硬化有关,是假说本身的问题,还是流行病学研究设计的问题,对此存在不同观点。
正方观点:感染是诱发动脉粥样硬化、导致斑块破裂的元凶
理论基础
动脉粥样硬化的发生发展与体内病原体反复或持续感染有关的学说已提出近百年。支持这一学说最直接的证据是从动脉粥样硬化斑块中检测出病原微生物;其次,用病原微生物感染实验动物可诱发出与人类相似的动脉粥样硬化病变。研究发现这些病原微生物与动脉粥样硬化发生、发展及动脉粥样硬化斑块的稳定性之间存在着密切联系,且对斑块的形态也产生一定的影响。
病原体可以直接感染血管壁细胞,并在细胞内潜伏,维持一个比较低水平的繁殖,也可能是间歇性地繁殖。病原体也可以被血液中的单核细胞携带到血管壁,能够感染单核细胞的病原体包括CMV、CP。CMV主要感染骨髓中单核细胞的前体。骨髓因此有可能成为CMV的感染灶。因此,这些被感染的骨髓中的单核细胞前体成为“特洛伊木马”,将CMV通过进入血液循环的单核细胞带到机体受损伤或者发生炎症的部位。尽管CMV在单核细胞中处于静止状态,一旦被单核细胞通过循环带到机体受损伤或者发生炎症的部位,单核细胞转化为巨噬细胞,CMV开始在血管壁的内皮细胞和平滑肌细胞表达即刻早期病毒基因产物(immediate early viral gene product)。
处于血管壁上的CMV,HSV和CP可以产生促动脉粥样硬化作用,如平滑肌细胞的增殖和迁移,增加细胞因子、趋化因子和细胞黏附分子的表达,增加活性氧的生成。CMV感染的平滑肌细胞可以增加氧化型低密度脂蛋白的吸收。
此外,血清流行病学证实与人类动脉粥样硬化有关的病原体可以在细胞内长期存在而并不繁殖,这种现象叫做不全感染(Abortive Infection)。但仍然能够通过病原体介导的分子机制抑制凋亡途径,促进动脉粥样硬化的发生。比如,CMV产生的即刻早期病毒基因产物——IE2-84能够抑制P53的转录活性,而P53是启动细胞凋亡从而抑制细胞增殖的重要基因。通过此途径,CMV促使被感染细胞的增殖。也就是说,CMV可以引起平滑肌细胞的不全感染,促使平滑肌细胞的增殖和迁移,动脉发生粥样硬化。
已发现动脉粥样硬化病变中含有大量的巨噬细胞和T淋巴细胞,提示病变内发生免疫应答反应(图1)。 C-反应蛋白(C-reactive protein, CRP)是一种反映全身低度炎症的非特异性标志物。有报道,约75%血清CMV感染阳性者有CRP水平升高;血清CMV感染阳性和CRP升高者经冠状动脉影像确诊为冠心病的可能性最大,提示CMV感染与冠心病相关联。
实验研究
体外及在体动物实验表明,感染可以导致血管内皮损伤、细胞因子分泌增加、高凝状态等促动脉粥样硬化的环境。冠状动脉粥样硬化的发生发展可能不仅与一种病原体感染相关,而是多种病原体共同感染并相互作用的结果。多种病原体感染可能通过一系列生物化学级联反应促使炎症、动脉粥样硬化、冠状动脉粥样硬化斑块破裂和血栓形成,进而导致冠心病发生发展。感染负荷可作为冠心病严重程度的预测指标之一。 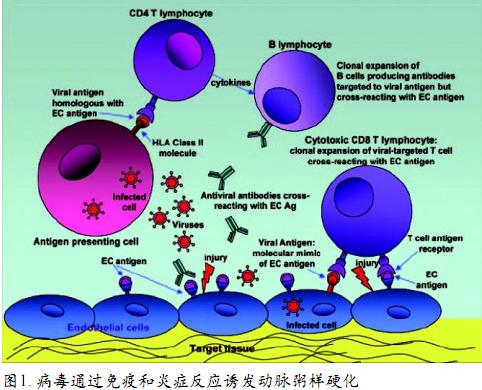
在芬兰及加拿大进行的两项研究中,在兔鼻内接种肺炎衣原体后出现主动脉粥样硬化。接种2次者较接种1次者发生率高,而假接种的对照组或感染支原体的对照组并无主动脉粥样硬化发生。Muhlestein等在胆固醇诱导的主动脉粥样硬化模型兔中分3次接种肺炎衣原体,3个月后主动脉病变明显加重。而在第3次接种后给予阿奇霉素治疗7周者,主动脉病变与对照组相似,并没有加重,治疗与不治疗者病变程度明显不同。Grayston给动脉粥样硬化实验鼠鼻内接种衣原体后发现,衣原体在主动脉壁上至少存在20周,病变中发现有衣原体,而且感染鼠与对照鼠比较,病变范围明显增大,16周时即已较对照鼠大1倍。上述研究结果表明,肺炎衣原体能够引起动脉粥样硬化,并可使病变过程加速。对机制的研究发现,体外培养的人巨噬细胞能够摄入一定量的脂类物质,如果细胞感染了肺炎衣原体,脂质摄入量更大。许多受感染的巨噬细胞在形态学上都极似粥样硬化斑块中的泡沫细胞。进一步研究发现,粥样硬化病变中存在有调节巨噬细胞的肿瘤坏死因子及基质金属蛋白酶表达的衣原体休克蛋白,其作用与动脉粥样硬化及其并发症有关。有人发现,内皮细胞感染衣原体后,对中性粒细胞及单核细胞的通透性增强,促进其在内皮下聚集。 
临床研究
早在20世纪初,美国和欧洲的流感暴发后,就有数据显示流感与冠心病及心肌梗死之间可能存在关联性。一项来自德国的研究为“感染负荷(infectious burden)与动脉粥样硬化的进展有关”这一观点提供了证据(图2)。 该研究对572例住院接受冠状动脉造影的患者分别测量了4种病毒及4种细菌株的IgG 或 IgA抗体,并随访3年。研究发现,患者对病原体产生的抗体种类越多,出现进展性动脉粥样硬化的可能性越大,死亡的危险性也越高。在已经患有进展性冠心病的患者中,这一关系尤为明显。在患有进展性动脉粥样硬化的患者中,如对0~3种病原体产生抗体,其死亡率为7.0%;对6~8种病原体产生抗体,死亡率增至20.0%。调整年龄、性别、心血管病的危险因素及CRP水平后,4种病原体抗体与动脉粥样硬化的严重程度之间仍然存在明显的关系。研究证实,感染总负荷与动脉粥样硬化的严重程度有关。
在动脉粥样硬化因果关系的研究中肺炎衣原体论据最多。血清流行病学、病理学和动物模型研究资料均反映其与动脉粥样硬化的病因学关系。离体和在体实验研究表明,肺炎衣原体作用的病理机制可能是加速局部脂质沉积。还有研究表明,几乎半数的动脉粥样硬化斑块中存在肺炎衣原体及肺炎衣原体产物,如热休克蛋白60及脂多糖,可激活巨噬细胞并促进血管壁炎症反应,说明肺炎衣原体可能促进斑块进展。
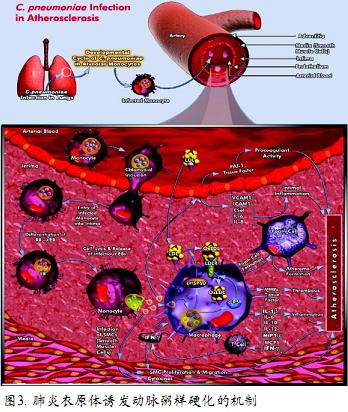
研究人员早就发现,人的呼吸道感染肺炎衣原体,心肌缺血的危险提高2~4倍,衣原体常与动脉粥样硬化斑块一起位于血管内膜。一项对死于心肌缺血患者的血管或手术中切除的动脉粥样硬化血管样本的研究发现,60%血管内膜脂质斑块处存在衣原体,其动脉粥样硬化更加显著。 进一步研究发现,血管内膜存在衣原体感染可损伤血管内膜,即使血液胆固醇含量正常,也可导致脂质斑块形成。脂质斑块和衣原体感染均可引发血管内膜的免疫和炎症反应,促使动脉粥样硬化发生和发展。如果血液中胆固醇含量过高,免疫性炎症过程将大大加速(图3)。 总之,感染与动脉粥样硬化之间有明确的关系,动物实验及临床试验进一步予以证实。
反方观点:感染未必诱发动脉粥样硬化、导致斑块破裂
理论基础
动脉粥样硬化是一种多因素性疾病,受到血压、血脂、血糖等多种危险因素,以及遗传等的影响,难以确定某一可能因素的致病作用。同样,感染引起的慢性炎症反应与动脉粥样硬化的关系也很难确定,主要在于涉及的因素较多。其一,病原体的种类与性质,宿主与致病物之间相互作用,这是决定病原体是否影响动脉粥样硬化过程的一个重要因素。其二,外周血单核细胞、巨噬细胞和T淋巴细胞分泌的多种细胞因子间相互作用的参与,以及与脂代谢及遗传背景[下一页] [1] [2] [3]